High Cholesterol And Heart Disease — Myth or Truth?
The Response-to-Injury Rabbit Never Developed Atherosclerosis — Why Not?August 23, 2008
by Chris Masterjohn
The pop science version of cholesterol goes something like this: when you eat fatty foods, especially foods rich in animal fat, the saturated fat and cholesterol in these foods wind up in your blood and stick to your arteries. Since saturated fats are solid outside your body, they will be solid inside your body too — depsite the 30-degree increase in average temperature. Arteries are much like pipes. When they get caked up with grease, blood flow is impaired, and a heart attack ensues.
None of the prominent scientists who promoted the idea that cholesterol is a critical factor in the development of heart disease ever believed anything remotely resembling this nonsense. From the beginning, they recognized that atherosclerotic plaque accumulates behind the layer of the artery in contact with the blood, called the endothelium, and that the cholesterol and fat within it is engulfed in white blood cells.
The theory these scientists promoted looked something like this:
when the cholesterol level in the blood increases, it penetrates the arterial wall and gets stuck;
- white blood cells circulating in the blood then enter the arterial wall and gobble up the cholesterol;
- the accumulation of lipid-loaded white blood cells causes local injury, leading to cell death, calcification, and the development of a collagen-laden "fibrous cap" over the atherosclerotic lesion;
- when the cap ruptures, the blood clots, blocking the artery and causing a heart attack.
But is this true? Books and web sites devoted to debunking this theory have come out of the woodwork over the last decade; books defending it have followed suit. Consider the following titles to see just how controversial the idea really is:
- The Cholesterol Myths: Exposing the Fallacy That Saturated Fat and Cholesterol Cause Heart Disease by Uffe Ravnskov, MD, PhD (2000).
- The Great Cholesterol Con: Why everything you've been told about cholesterol, diet, and heart disease is wrong! by Anthony Colpo (2006).
- The Cholesterol Wars: The Skeptics vs. the Preponderance of the Evidence by Daniel Steinberg, MD, PhD (2007).
- The Great Cholesterol Con: The Truth About What Really Causes Heart Disease and How to Avoid It by Malcolm Kendrick, MD (2007).
So is the theory that cholesterol causes heart disease just a myth? Or are the skeptics truly waging a war against the preponderance of the evidence?
In this Article:
- The Cholesterol Debate — What Causes Atherosclerosis?
- The Response-to-Injury Rabbit Model
- Atherosclerosis is Just One Type of Arteriosclerosis
- Medial Calcification and the Vitamin K2 Connection
- The Cholesterol-Fed Rabbit Controversy
- Cholesterol in the Blood, Not the Food
- Lessons From Familial Hypercholesterolemia
- The Role of Oxidized LDL in Heart Disease
- The Discovery of Oxidized LDL
- Oxidized LDL Causes Injury and Inflammation
- Small, Dense LDL and Oxidation — Which Causes Which?
- Oxidized LDL and the PUFA Connection
- Shear Stress Explains the Locations of Plaques and the Benefits of Exercise
- What About Correlations With High Cholesterol?
- So is the Lipid Hypothesis Correct?
- References
The Cholesterol Debate — What Causes Atherosclerosis?
The truth is that each of these authors makes important points. Were there never any good evidence that cholesterol was involved in heart disease, there would be no National Cholesterol Education Program, no statin empire, and Daniel Steinberg could never have written a book plus over 200 scientific papers on the subject. On the other hand, were there never anything seriously wrong with the mainstream dogma on the issue, Ravnskov, Colpo, Kendrick, and many other authors could never have built their careers around pointing out the gaping holes in the theory.There is no one cause of "heart disease." "Heart disease" is a heterogeneous compliation of diseases of the heart and blood vessels with many different causes. Some of these include disturbances of the rhythm of the heart, calcification of the middle portion of the blood vessels and calcification of the heart valves, and congestive heart failure. The question I address in this article is whether and in what sense cholesterol is involved in atherosclerosis, the development of fatty and calcified plaques in isolated, raised lesions, which can cause heart attacks by rupturing, clotting, and blocking arteries.
In 1933, the famous proponent of the cholesterol-fed rabbit model Nikolai Anitschkov declared that atherosclerosis had been shown to be of an "infiltrative" character rather than a "degenerative" character and was driven by lipids (fatty substances) rather than by inflammation. He did not deny inflammation was involved, but believed that it was secondary to lipid infiltration. Many opponents continue to claim that the root cause driving heart disease has nothing to do with lipids and everything to do with inflammation and that it is degenerative rather than infiltrative in character.
As we will see below, these are all correct! Atherosclerosis is largely driven by the degeneration of lipids which infiltrate the blood vessel and thereby cause inflammation. Inflammation from other sources may accelerate the process or further the degeneration of the atherosclerotic plaques once they are formed, but the initiating factor for fatty plaques appears to be the degeneration of lipids — especially the degeneration of polyunsaturated fatty acids (PUFA).
In order to begin looking at the evidence, we must go back a century in time to the cholesterol-fed rabbit. The cholesterol-fed rabbit model came on the heels of extensive investigations into what would later be termed the "response-to-injury hypothesis."
The Response-to-Injury Rabbit Model
Around the turn of the twentieth century, research into the cause or causes of heart disease was in full throttle. A 1933 compilation edited by E.V. Cowdry entitled Arteriosclerosis: A Survey of the Problem (New York: Macmillan) contained twenty reviews of investigations into the matter, including statistical relationships, the distribution of the disease in wild animals, the distribution in humans according to race and climate, nutritional influences, the physical and chemical nature of the changes that occur in atherosclerotic tissues, and experimental models of the disease.Nikolai Anitschkov, who developed the cholesterol-fed rabbit model, wrote the 50-page review of experimental animal models.1 Much of this research was published in German, so Anitschkov's review is an invaluable resource.
According to Anitschkov, early ideas about the origin of arteriosclerosis — a general term for hardening and damage to the arteries, of which atherosclerosis is a specific type — saw the diseases as a response to injury. The injury was primarily seen as either a mechanical or a toxic factor, and was sometimes believed to be injury to the nerves rather than injury to the blood vessels. Researchers carried out a multitude of experiments on rabbits and other animals, including the following:
- Mechanical damage to the blood vessels including ligating, pulling, pinching, and wounding them, and cauterizing them with galvanic wire or silver nitrate.
- Increasing blood pressure by constricting the blood supply through the aorta, damaging the kidneys, or hanging rabbits up by their feet.
- Severing or irritating certain nerves.
- Injecting rabbits with adrenalin.
- Injecting rabbits with a multitude of toxic factors, including digitalin, strophanthin, adonidin, ergotin, theocin, barium chloride, hydrastin, nicotine, caffeine, formalin, ergosterol, and various salts of acids and heavy metals.
- Injection of diphtheria toxin and many other bacteria cultures or bacterial byproducts.
Atherosclerosis is Just One Type of Arteriosclerosis
None of these methods, however, produced anything resembling human atherosclerosis. While arteriosclerosis refers to hardening and degeneration of the arteries in general, atherosclerosis is a specific type of arteriosclerosis in which a plaque rich in lipid-loaded white blood cells, cholesterol, fatty acids, calcium, various debris — called an atheroma — invades the innermost layer of the blood vessel wall called the intima, just behind the one-cell-thick layer called the endothelium. If you are not familiar with the anatomy of a blood vessel, you can see a diagram of it here.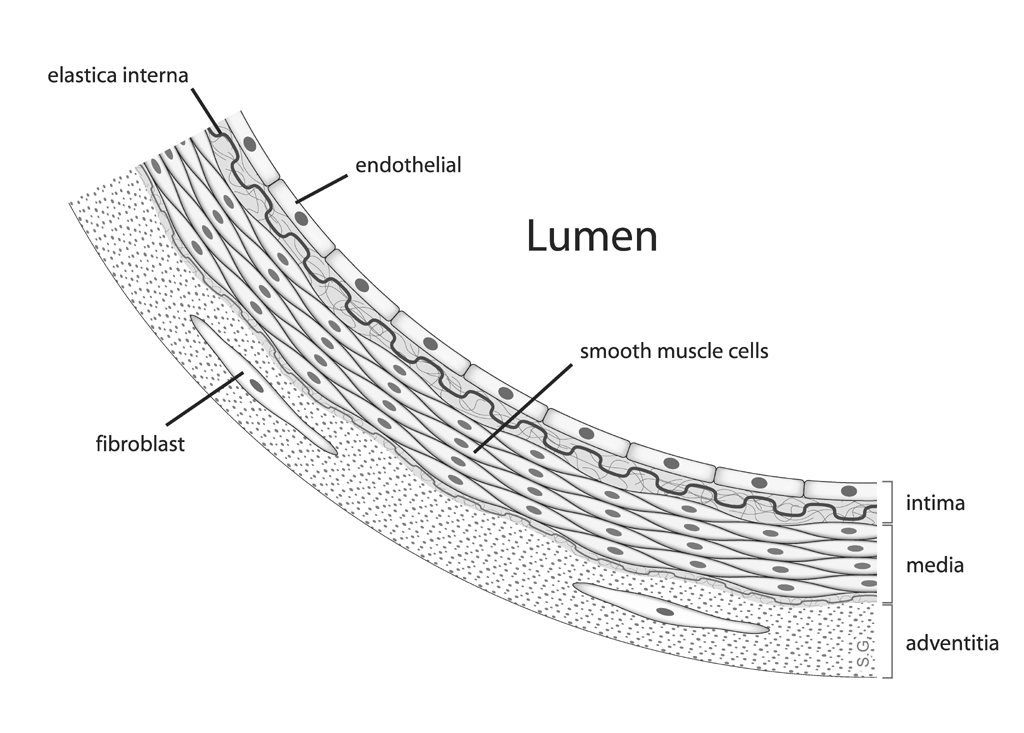{kind=link}
The research in Anitschkov's day suggested that, while various types of arteriosclerosis occurred in humans, atherosclerosis was a much more important cause of death. Anitschkov thus concerned his research with what caused atherosclerosis.
The mechanical injuries to blood vessels or nerves produced a local repair process that involved the proliferation of cells, their congregation around the damaged area, and a resultant thickening of the vessel wall. The results were local rather than systemic, however, and never produced a lesion resembling an atheroma.
Injections of adrenalin produced much more interesting changes that were much more relevant to humans. They produced necrosis (death) of cells in the media followed by extensive calcification. A similar process was observed in some of the blood pressure experiments and in many of the experiments involving injections of metallic, bacterial, or other toxins. These changes, however, were fundamentally different from atherosclerosis, which occurs in the intima.
Medial Calcification and the Vitamin K2 Connection
That does not mean this research is irrelevant. Humans experience this type of medial calcification in diabetes, kidney disease, and aging. It appears to assault the media of arteries and the valves of the heart together. It increases arterial stiffness and decreases the artery's ability to accomodate moderately high levels of blood pressure. One of the most important factors in this type of calcification appears to be vitamin K2.Vitamin K-dependent proteins protect against cell death, help clear away the debris that cells leave behind when they do die, and protect against the calcification of soft tissues. In the absence of sufficient vitamin K, these proteins are deformed and fail to work properly. It appears that vitamin K2, found in animal fats and fermented foods, is far more important in this respect than vitamin K1, found in green plant foods. I have written extensively on this subject and argued that vitamin K2 is the "activator X" of Weston Price in my article, On the Trail of the Elusive X Factor: Vitamin K2 Revealed.
Despite the research in Anitschkov's day suggesting that only atherosclerosis had major clinical importance, research in our own day shows that calcification of the media and valves is critically important to, at a minimum, the 324 million people worldwide who will be diabetic come 2025. For the US population born in 2000, the estimated lifetime risk of type 2 diabetes is one in three.2 In type 2 diabetics, medial calcification increases the risk of mortality from heart disease, stroke, and all causes. It also predicts the incidence of heart disease and stroke, including events that do not produce fatalities, and predicts the likelihood that peripheral artery disease will require limb amputation.3
So the response-to-injury hypothesis has a solid basis of evidence for arteriosclerosis of the media, and this is clinically important — but what causes atheroma, that is, the fatty plaque that causes raised lesions in the intima of the blood vessels?
To answer this question, we must look to the cholesterol-fed rabbit.
The Cholesterol-Fed Rabbit Controversy
In 1909, a researcher at the Military Medical Academy in St. Petersburg named Ignatowski produced atherosclerosis in rabbits by feeding them a diet of meat, eggs, and milk. He was pursuing a hypothesis put forward by Nobel Prize-winning microbiologist I. Metchnikov that dietary protein accelerated aging.4In 1913, Anitschkov and his partner Chalatov were studying at the same academy and were assigned to follow up Ignatowski's work. They progressively narrowed down the causative factor to cholesterol by feeding different foods and fractions of foods, finally producing the diease by feeding pure cholesterol dissolved in sunflower oil.4
Rabbits fed sunflower oil alone did not develop atherosclerosis. In the cholesterol-fed rabbits, however, lesions developed that exhibited a remarkable similarity to the human disease. They began as fatty streaks in the intima; circulating white blood cells then invaded the intima and engulfed the cholesterol and fat deposited there, eventually growing into large phagocytic cells that Anitschkov called xanthoma cells and we now call foam cells; eventually the developing plaque protruded into the intima in the form of a raised lesion. The lesion possessed a fatty core rich in crystalized and calcified cholesterol deposits and was covered with a fibrous cap.1
The lesions did not appear everywhere equally, but occurred in specific areas. They were most prominent in the aorta and other large arteries, especially in the areas of the artery wall that experience disturbed blood flow such as the points where the arteries branch. While they did not occur in exactly the same places as human atherosclerotic lesions, the pattern was largely similar and the underlying physiological principle dictating the location of the lesions — mainly the type of blood flow experienced by the artery wall — was the same.1
The rabbits developed cholesterol deposits all throughout their bodies, in their eyes and internal organs. Anitschkov produced a more mild form of the disease, however, by feeding the rabbits milk. In these experiments, the rabbits received a much more moderate amount of cholesterol over a much longer period of time and the resulting disease was much more focused in the arteries.1
One curious difference between rabbits and humans is that when rabbits develop atherosclerosis, their plaques never rupture and they never get heart attacks. The main determinant of plaque rupture according to the current scientific literature is the balance between collagen degradation and collagen synthesis.5 Collagen synthesis requires vitamin C. Most animals, including rabbits, make their own vitamin C, but humans do not.
Atherosclerosis itself probably diminishes the quality of life in many different ways by impeding blood flow and blood vessel function, but it clearly does not inexorably lead to heart attacks. The reason why atherosclerosis produces heart attacks in humans and not rabbits or many other animals might be that humans cannot produce their own vitamin C.
Cholesterol in the Blood, Not the Food
Anitschkov argued against calling cholesterol "the cause" of atherosclerosis, but he considered cholesterol the primary causal factor and the necessary causal factor. Mechanical injuries, adrenalin injections, and other methods used to induce various types of arteriosclerosis would accelerate the development of atheroma when they were combined with cholesterol-feeding, but they would never result in human-like atherosclerosis by themselves.Anitschkov never concluded from his experiments that cholesterol in the diet caused atherosclerosis in humans, however. To the contrary, he wrote the following:
[I]n human atherosclerosis the conditions are different. It is quite certain that such large quantities of cholesterin are not ingested with the ordinary food. In human patients we have probably to deal with a primary disturbance of the cholesterin metabolism, which may lead to atherosclerosis even if the hypercholesterinemia is less pronounced, provided only that it is of long duration and associated with other injurious factors.Cholesterol skeptics often argue that the rabbit is irrelevant to the human because it is an herbivore. Cholesterol-feeding has failed to produce atherosclerosis in many other species. This is true, but it misses the point. In the species where cholesterol-feeding alone does not produce atherosclerosis, the blood level of cholesterol does not rise as much as in rabbits. But in all of these species when the level of cholesterol in the blood rises high enough, atherosclerosis ensues. For example, feeding dogs cholesterol alone does not produce atherosclerosis because they turn the cholesterol into bile acids; but inhibiting thyroid hormone stops them from making this conversion, and when combined with cholesterol-feeding, it induces atherosclerosis.
As Steinberg points out, raising blood levels of cholesterol has produced atherosclerosis in baboons, cats, chickens, chimpanzees, dogs, goats, guinea pigs, hamsters, monkeys, mice, parrots, pigs, pigeons, rabbits and rats.
The role of blood cholesterol in human heart disease was supported by research showing that people with a disorder that would eventually be called familial hypercholesterolemia had dramatically increased blood levels of cholesterol and, in youth and middle age, dramatically increased relative risks of heart disease and atherosclerosis. But what caused their high cholesterol levels, and did those levels cause the atherosclerosis, and if so, did this phenomenon have any relevance to the rest of us?
And, if cholesterol was somehow the culprit in all of this, was it merely its concentration in the blood that was at play, or was something very different going on?
Lessons From Familial Hypercholesterolemia
Familial hypercholesterolemia (FH) bears a striking resemblance to the cholesterol-fed rabbit model. In mild cases, it produces earlier and more rapidly developing atherosclerosis compared to the general population. In its severe cases, it results in cholesterol deposits all throughout the body, especially in the liver, kidneys, and eyelids.4In the mid-1970s, Brown and Goldstein discovered that FH resulted from a single genetic defect in the LDL receptor that made the cells unable to absorb LDL from the bloodstream. Steinberg argues that, since cells jealously guard their cholesterol concentrations by adjusting their synthesis of cholesterol as needed, this showed that FH patients differed from the general population in only one single way: the concentration of cholesterol in their blood.4
The finding drew several more parallels between FH and Anitschkov's cholesterol-fed rabbits. Anitschkov argued that it was not the mere feeding of cholesterol to the rabbits that produced atherosclerosis, but the overwhelming of their capacity to use and dispose of that cholesterol. FH cells could absorb free cholesterol, but not cholesterol from LDL. Anitschkov's rabbits developed atherosclerosis when they ate cholesterol, but not when they were injected with it — in which case it would not be packaged into lipoproteins such as LDL, which contain many other substances besides cholesterol. Looking backward, it appears that the common thread running through each model was that the level of LDL in the blood exceeded the capacity of the LDL receptors to move that LDL from the blood to the cells.
The LDL receptor highway was blocked, and the LDL traffic was jammed.
Is Steinberg correct, however, that this changes nothing but the concentration of LDL in the blood? Consider what happens in a traffic jam:
- The concentration of cars in the road increases.
- It takes you longer to get home.
The Role of Oxidized LDL in Heart Disease
Anitschkov believed that his research showed that atherosclerosis was of an infiltrative character rather than a degenerative character. He believed that cholesterol and other substances naturally permeated the endothelium in order to nourish the other layers of the blood vessels, and proceeded from there into the lymph fluid. When the blood level of cholesterol rose sufficiently, he argued, it entered the intima at a faster rate than it could exit and began to accumulate.Anitschkov was correct that the disease was driven by an infiltration of lipid, and he was correct that the degeneration of the blood vessel wall was secondary to this infiltration. What he failed to realize, and could not have realized at the time, was that the entire process depended on the degeneration of the lipid.
The Discovery of Oxidized LDL
Beginning in 1979, investigators made a series of revolutionary discoveries revealing this degenerative process. When they incubated cells with LDL in the absence of other serum components, the cells underwent severe damage and began to die within 24 hours. Adding serum or HDL prevented the toxicity.4In 1981, these researchers discovered that culturing endothelial cells with LDL caused dramatic changes to the LDL, making it denser, more electronegative, and giving it a dramatic ability to accumulate in white blood cells called macrophages. Macrophages are phagocytic, meaning they like to gobble up other things, and they are the precursors to the "foam cells" that populate atherosclerotic plaques. The researchers called this LDL "endothelial cell-modified LDL." Soon after, they discovered that the LDL was being "oxidatively modified" and that not only HDL but vitamin E (which HDL is rich in) prevented the effect.4
Oxidized LDL Causes Injury and Inflammation
Since those early findings, thousands of papers have now been published on the role of oxidized LDL in the development of atherosclerosis. Oxidized LDL causes endothelial cells to secrete "adhesion molecules" and "chemoattractants" that allow white blood cells called monocytes to penetrate in between the endothelial cells and stick to them in the subendothelial space where fatty streaks and atherosclerotic plaques develop.6Oxidized LDL turns on the expression of genes in monocytes which cause them to convert into macrophages and eventually into foam cells, which makes them gobble up more and more oxidized LDL endlessly — but these macrophages use "scavenger receptors" rather than LDL receptors, so they never take up meaningful amounts of non-oxidized LDL; they only take up oxidized LDL, and it is oxidized LDL itself that initates this endless cycle.7
Oxidized LDL initiates the inflammatory process by causing foam cells to secrete molecules that attract T cells and other inflammatory cells.6 Oxidized LDL enhances the process whereby T cells, foam cells, smooth muscle cells and endothelial cells decrease collagen production and increase collagen degradation, which leads to the rupture of the fibrous plaque.5
Endothelial cells produce nitric oxide, a gas that protects LDL from oxidation, increases blood flow, decreases the adhesion of monocytes to the endothelium, and decreases blood clotting. Oxidized LDL impairs the endothelial cell's ability to produce nitric oxide.8
In short, oxidized LDL contributes to the entire atherosclerotic process from start to finish. Writers who argue that atherosclerosis has nothing to do with lipids but is all about inflammation and response to injury must contend with the fact that oxidized LDL injures endothelial cells and causes inflammation!
Small, Dense (Pattern B) LDL and Oxidation — Which Comes First?
If it is oxidized LDL rather than LDL per se that contributes to atherosclerosis, the question arises of what causes LDL to oxidize. Since polyunsaturated fatty acids (PUFA) in the LDL membrane are the components that are most vulnerable to oxidation, excess PUFA and insufficient antioxidants would seem to be the most obvious culprits. Endothelial cells, however, secrete a number of oxidative enzymes such as myeloperoxidase and lipoxygenase. LDL is always exposed to endothelial cells in the blood, but if it makes its way into the subendothleial space where it can get stuck in a network of sugary proteins called proteoglycans, it would be exposed to them even more directly. Some researchers have therefore put forward the "response-to-retention hypothesis," wherein the LDL oxidizes in response to getting stuck in the subendothelial space.In 1988, a case-control study showed that people with a preponderance of small, dense LDL were three times more likely to suffer from a heart attack.9 Researchers subsequently showed that the smaller and denser LDL gets, the more quickly it oxidizes when they subject it to oxidants in a test tube.10 Then the "response-to-retention" crowd jumped in on the game a few years later and showed that small, dense LDL were much more likely get stuck in test tube versions of the proteoglycan network of the subendothelial space.11
If the response-to-retention hypothesis is true, we are back to the infiltration hypothesis where the accumulation of LDL in the subendothelial space is driving the whole process because the accumulation causes the oxidation. This would be a convenient way of circumventing the enormously embarassing fact that the medical establishment has been pushing highly oxidation-prone PUFA oils for fifty years.
The question is, how are these LDL getting small and dense?
Within the response-to-retention paper, the authors stated that "with decreasing size and increasing density the LDL particles have less of the non-polar core covered with a surface monolayer made of phospholipids and cholesterol."
Where did the phospholipid membrane go?
A group working on lipoprotein (a), or Lp(a), published a paper in July of this year showing that virtually all oxidized LDL in the blood circulates attached to Lp(a). Lp(a) is essentially LDL stuck to a protein called apolipoprotein (a) or apo(a). This group showed that when oxidized LDL and apo(a) are incubated together, many of the oxidized phospholipids transfer directly to the apo(a).12 In other words, when the membrane of LDL begins to oxidize, parts of it hop right off the LDL particle. Could that explain why "less of the non-polar core" would be "covered with a surface monolayer" on some LDL particles?
When Steinberg and his coworkers first described the characteristics of "endothelial cell-modified LDL," one of the most conspicuous changes that occurred to the LDL particles was a marked increase in density.13
A 1997 study confirmed that the LDL taken from people with a preponderance of the small, dense type does indeed oxidize quicker in a test tube, but the oxidation status of the LDL was different before they subjected it to oxidation. The predominantly small, dense LDL had a higher ratio of oxidized-to-reduced coenzyme Q10 and a lower CoQ10-to-vitamin E ratio.14 Since CoQ10 is the first line of defense against LDL oxidation, this study strongly suggested that oxidation of the small, dense LDL had already started.
So here we have a chicken-and-egg question. Does small, dense LDL oxidize more rapidly in a test tube because it is small and dense, or because it is already partially oxidized, and its antioxidant defenses are already partially depleted? Is small, dense LDL more vulnerable to oxidation, or does LDL become small and dense when it becomes oxidized?
If LDL becomes small and dense through oxidation, then even if the test tube studies on its "stickiness" are correct and small, dense LDL is more likely to get stuck in the sugary protein network behind the endothelium, it is the oxidation driving the stickiness and not the stickiness driving the oxidation.
So we are back to square one wondering why the medical establishment never announed an emergency measure to put all the research dollars into discovering just how much damage it had done to everyone who followed its recommendations to use high-PUFA vegetable oils in place of saturated animal fats over the last fifty years.
Oxidized LDL and the PUFA Connection
Let us return to the traffic analogy for a moment. Why would an "LDL traffic jam," wherein the "LDL receptor highway" is blocked contribute to atherosclerosis?The membrane of LDL contains polyunsaturated fatty acids (PUFA), which are highly vulnerable to oxidation. Cells continuously make antioxidant enzymes and other antioxidant compounds to protect their membrane PUFA. If PUFA start to oxidize, the cell ramps up its antioxidant production. When the liver packs cholesterol into a VLDL particle and secretes it into the blood (where it eventually becomes an LDL particle after delivering some of its nutrients to other tissues), it puts some antioxidants into the package. The PUFA have now left the comparative safety of the liver cell and have only a limited supply of antioxidants. When those antioxidants are used up, the PUFA begin to oxidize, and their oxidation products proceed to damage other components of the lipoprotein. When the oxidation becomes severe, the oxidized LDL winds up in a foam cell in an atherosclerotic plaque.
Let's draw another analogy, this time to a jar of oil. If you use a jar of oil, you open it, exposing the PUFA within it to the oxygen in the air, but quickly put the cap back on and put it back in the fridge. What would happen if you opened the jar and let it sit on the table at room temperature? Over time, the limited amount of antioxidants in the oil would run out and the PUFA would begin to oxidize. The oil would go rancid.
Pumping LDL into the blood but letting it sit there circulating round and round exposed to oxidants rather than taking it into the shelter of the cell is like opening a jar of oil and leaving it on the table.
LDL taken from people who consume more PUFA, whether from vegetable oil or fish oil, oxidizes more easily in a test tube. Alpha-tocopherol, the major form of vitamin E, does not help.15
The specific components of the oxidized LDL particle that interact with the DNA of monocytes to transform them into macrophages and then into foam cells are oxidized derivatives of linoleic acid, a PUFA found in vegetable oils.16
A 2004 study from Brigham and Women's Hospital and Harvard School of Public Health showed that in postmenopausal women, the more PUFA they ate, and to a much lesser extent the more carbohydrate they ate, the worse their atherosclerosis became over time. The more saturated fat they ate, the less their atherosclerosis progressed; in the highest intake of saturated fat, the atherosclerosis reversed over time.17
I will cover the topic of saturated fat, PUFA, and heart disease in greater detail in another article on the diet-heart hypothesis. Additionally, I have written a Special Report entitled How Essential Are the Essential Fatty Acids? that provides accurate and thoroughly researched information on the true requirement for PUFA, which is negligible for healthy adults. As part of my Special Reports series, I will be publishing a second PUFA Report later this year that will cover the benefits and dangers of consuming PUFA in amounts larger than the minimum requirements.
Shear Stress Explains the Locations of Plaques and the Benefits of Exercise
As in the cholesterol-fed rabbit, human atherosclerosis occurs in discrete plaques at specific locations. In both species, these plaques occur in locations that experience disturbed blood flow, such as the points where the arteries branch.Anitschkov showed that the endothelium was more permeable to molecules labeled with dye at these points. Experimental vessel injuries that caused inflammatory responses made the endothelium even more permeable, but even in the absence of any treatment, the endothelium was naturally permeable in these areas.1
Sections of the arterial wall in these areas experience a lower level of shear stress than sections in other areas. Shear stress is the type of pressure that is caused by laminar blood flow, or the flow of blood parallel to the blood vessel wall. Shear stress decreases the permeability of the endothelium by stimulating the production of the proteins that form the junctions between the endothelial cells. Under levels of shear stress approximating those that exist at locations where atherosclerosis develops, easily visualizable gold particles the size of LDL particles slip right in between the endothelial cells, whereas the permeability to these particles is very low under levels of shear stress approximating those that exist where plaques do not develop.18
Shear stress also increases nitric oxide production. Nitric oxide increases blood vessel dilation and blood flow, decreases the adhesion of monocytes to the endothelium, decreases blood clotting, and prevents the oxidation of LDL.6
By increasing blood flow, exercise increases shear stress. Since the average shear stress over time seems to be the critical factor, exercise might help prevent atherosclerosis by decreasing the permeability of the endothelium and increasing nitric oxide production in those areas of the blood vessels where the resting level of shear stress is insufficient for protection.
What About Correlations with High Cholesterol?
Much of the cholesterol debate focuses on correlations with cholesterol. How strong are they? How consistent are they? Why do they show up in young people but not in old, in men more than women?The debate really misses the point, because since the early 1980s the molecular evidence has made very clear that it is oxidized LDL that contributes to atherosclerosis.
Correlations with cholesterol are likely to be confounded by a variety of factors that simultaneously increase cholesterol levels and contribute to heart disease, like stress and inflammation. In fact, inflammation seems to increase cholesterol synthesis essentially as an accidental byproduct of activating the stress response through an enzyme called Rho. Rho slashes nitric oxide production and thus almost certainly makes a contribution to atherosclerosis. For more information on Rho activation, click here.
Researchers have only recently developed methods for testing levels of oxidized LDL. One group has developed an antibody that recognizes oxidized but not non-oxidized phospholipids. They have shown that the proportion of LDL-associated phospholipids that are oxidized is a much more impressive risk factor for heart disease than LDL, and when it is multiplied by the level of LDL, thus indicating the total concentration of oxidized phospholipids, it is even better. Its predictive value is lower in older people, but still strong.19
Why would the association decline with age? If we look at the totality of the evidence about the mechanisms of atherosclerosis, it appears that oxidized LDL is the necessary initiating factor, but that we should expect its prominence as a contributing factor to decrease over time. Atherosclerosis probably does not develop in the absence of oxidized LDL. Once it does develop, however, and once the oxidized LDL stimulates the formation of foam cells, those foam cells recruit T cells that make their own contribution to the inflammatory process. Animal experiments show that independent sources of inflammation cannot initiate atherosclerosis, but they can aggravate it or accelerate it. Vitamin C deficiency, systemic infection, stress, and many other factors likely make contributions alongside oxidized LDL to the weakening and rupture of the fibrous cap that ultimately leads to a heart attack.
Virtually everyone develops substantial atherosclerosis by the time they are old. In people with more oxidized LDL, it occurs faster, and consequently reaches an advanced stage at a younger age. Inflammation will not help rupture a plaque that does not exist, so it will be much less likely to cause a heart attack in a younger person unless that person has high levels of oxidized LDL and consequently advanced atherosclerosis. In an older population wherein most people have advanced plaques, the factors that weaken the plaque will become much more important than the factors that create the plaque.
Studying the issue is complicated by the fact that we are looking at oxidized LDL in the blood. Once LDL gets oxidized enough, presumably it will wind up in arterial plaque. If there are factors that protect circulating LDL from contact with the tissues it could harm, they could confound the association.
Finally, studies looking at cardiovascular incidence or mortality are confounded by the fact that atherosclerosis is only one type of arteriosclerosis, and arteriosclerosis is only one cause of cardiovascular disease. Medial calcification, arrhythmia, congestive heart failure, or other causes of emboli (particles that can cause vessels) may all contribute to cardiovascular events. Oxidized LDL should only, or at least primarily, correlate with those events caused by atherosclerosis.
So is the Lipid Hypothesis Correct?
So is the lipid hypothesis correct? Not in its original form. The weight of the evidence clearly supports a role for the oxidation of LDL and not the concentration of LDL in the blood in the development of atherosclerosis.The oxidized lipid hypothesis has an enormous amount of evidence supporting it. The cholesterol-fed rabbit model was a model not merely of hypercholesterolemia but of hyper-oxidized-lipoproteinemia. Antioxidants cause major decreases in atherosclerosis in cholesterol-fed or Watanabe familial hypercholesterolemic rabbit models independent of cholesterol levels.4,20
We should not expect antioxidants to be fully capable of preventing the oxidation of LDL by themselves. As I discuss in my PUFA Report, antioxidants can stop oxidized PUFA from damaging other PUFA, but they can never fully repair the oxidized PUFA. The best they can do is convert it to a hydroxy-fatty acid, and it is the hydroxy versions of linoleic acid that have been shown to convert monocytes to foam cells!
Thus, all three of the following critical factors must be addressed:
- Increasing antioxidant status, especially coenzyme Q10, but also alpha- and gamma-tocopherol.
- Reducing PUFA intake.
- Increasing LDL receptor function to minimize the amount of time LDL spends in the bloodstream.
A recent study showed that curcumin, a component of tumeric, increases the expression of the LDL receptor. This study may provide valuable clues. Thyroid hormone is important to the function of the LDL receptor, and many people likely have suboptimal thyroid status.
The irony in all of this is that there is no evidence to suggest that cholesterol is the culprit. In the rabbit, consuming large amounts of cholesterol increases the exposure of LDL membrane-associated PUFA to oxidation because it causes their translocation from the liver to the blood where they are detached from the cellular environment and less protected. In humans, eating cholesterol in the form of several eggs per day probably decreases the vulnerability of LDL to oxidation. See here.
The higher the concentration of free cholesterol within the LDL particle, the less vulnerable it is to oxidation. By contrast, the higher the concentration of cholesterol that is linked to fatty acids, called "esterified cholesterol," the more vulnerable the LDL is to oxidation.9 Esterified cholesterol primarily exists in the core of the particle. Free cholesterol primarily exists in the surface membrane where the initial oxidation takes place, so cholesterol seems to protect the PUFA from oxidation.
So, does cholesterol cause atherosclerosis? No!
But do blood lipids? Yes. Atherosclerosis is a disease in which degenerating lipids infiltrate the blood vessel wall and cause inflammation and degeneration of the local tissue once they arrive there. Solid evidence has amassed in favor of this view for the last 100 years.
| Share this page: |  |  |  |  |
 |  |  |
References
1. Anitschkow N, Experimental Arteriosclerosis in Animals. In: Cowdry EV, Arteriosclerosis: A Survey of the Problem. 1933; New York: Macmillan. pp. 271-322.2. Cheng D. Prevalence, predisposition and prevention of type II diabetes. Nutrition & Metabolism. 2005;2:29.
3. Lehto S, Niskanen L, Suhonen M, Ronnemaa T, Laakso M. Medial Artery Calcification. A Neglected Harbinger of Cardiovascular Complications in Non-Insulin-Dependent Diabetes Mellitus. Arteriosis, Thrombosis, and Vascular Biology. 1996;16:978.
4. Steinberg D, The Cholesterol Wars: The Skeptics vs. The Preponderance of the Evidence.2000; San Diego: Academic Press.
5. Libby P. The molecular mechanisms of the thrombotic complications of atherosclerosis. J Intern Med. 2008;263(5):517-27.
6. Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83(suppl):456S-60S.
7. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241-52.
8. Laufs U, Fata VL, Plutzky J, Liao JK. Upregulation of Endotelial Nitric Oxide Synthase by HMG CoA Reductase Inhibitors. Circulation. 1998;97:1129-1135.
9. Austin MA, Breslow JL, Hennekens CH, Buring JE, Willet WC, Krauss RM. Low-density lipoprotein sublass patterns and risk of myocardial infarction. JAMA 1988;260(13):1917-21.
10. Tribble DL, Holl LG, Wood PD, Krauss RM. Variations in oxidative susceptibility among six low density lipoprotein subfractions of differing density and particle size. Atherosclerosis. 1992;93:189-99.
11. Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: Pathological significance and molecular basis. Atherosclerosis 1998;139:205-222.
12. Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin M-J, et al. A Novel Function of Lipoprotein (a) as a Preferential Carrier of Oxidized Phospholipids in Human Plasma. J Lipid Res. 2008 Jul 3;[Epub ahead of print]
13. Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: Recognition by receptors for acetylated low density lipoproteins. Proc Natl Acad Sci USA. 1981;78(10):6499-6503.
14. de Rijke YB, Bredie SJH, Demacker PNM, Vogelaar JM, Hak-Lemmers HLM, Stalenhoef AFH. The Redox Status of Coenzyme Q10 in Total LDL as an Indicator of In Vivo Oxidative Modification. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:127-133.
15. Nenseter MS, Drevon CA. Dietary polyunsaturates and peroxidation of low density lipoprotein. Curr Opin Lipidol. 1996;7(1):8-13.
16. Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93(2):229-40.
17. Mozaffarian D, Rimm EB, Herrington DM. Dietary fats, carbohydrate, and progression of coronary atherosclerosis in postmenopausal.
18. Conklin BS, Vito RP, Changyi C. Effect of Low Shear Stress on Permeability and Occludin Expression in Porcine Artery Endothelial Cells. World J Surg. 2007;31:733-43.
19. Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353(1):46-57.
20. Wang Z, Zou J, Cao K, Hsieh TC, Huang Y, Wu JM. Dealcoholized red wine containing known amounts of resveratrol suppresses atherosclerosis in hypercholesterolemic rabbits without affecting plasma lipid levels. Int J Mol Med. 2005;16(4):533-40.