Targeting insulin inhibition as a metabolic therapy in advanced cancer (Part 1 of 2)
Posted: October 15, 2012 in Cancer, Cell Signaling, ketogenic diet, low-carbohydrate dietTags: Cancer, carbohydrate, dietary guidelines, ketogenic diet, low carbohydrate
52
Guest post: Dr. Eugene J. FineDr. Feinman invited me to contribute a guest blog on our recently published cancer research study: “Targeting insulin inhibition as a metabolic therapy in advanced cancer: A pilot safety and feasibility dietary trial in 10 patients” which has now appeared in the October issue of the Elsevier journal Nutrition, with an accompanying editorial. Today’s post will focus on this dietary study, and its relation to the general problem of cancer and insulin inhibition. Part II, next week, will discuss in more detail, the hypothesis behind this study. Richard has already mentioned some of the important findings, but I will review them since the context of the study may shed additional light.
Comments to Richard’s post noted that almost 100 years ago Otto Warburg described how many cancers depend on glucose for fuel, and remarkably, that the cancers relied on anaerobic glycolysis for energy even under aerobic conditions. In contrast, normal skeletal muscle, for example, might rely on anaerobic metabolism in a sprint but would switch back to aerobic respiration (and reliance on fat) after slowing to a walk. The stubborn use of glucose and glycolysis by cancer cells has been termed “The Warburg effect.” The important point is that anaerobic metabolism relies on glucose and strictly glycolytic cells cannot use fatty acids for metabolism
Meanwhile, the 1982 Food Pyramid encouraged Americans to eat 300-400 grams of CHO per day. Many followed their advice, now to their regret, and at least 90% of these CHOs were sugars, or starches that digested to sugars, all useable by many cancers. So at first we thought we might starve tumors by limiting dietary carbohydrate (CHO), obviously a major source of blood glucose.
It took us less than a day of literature review to recognize that CHO restriction would not starve most cancers because they are usually excellent at pirating glucose at blood glucose concentrations way below the normal range.
A few words about me: I’ve been fascinated with the Warburg effect since medical school. My specialties are Internal Medicine and Nuclear Medicine. One of the radioactive isotopes that we use in Nuclear Medicine is Fluorine-18 (written 18F) which emits positrons. (Positrons interact with other substances to emit gamma rays, high energy light waves that mostly pass through tissues and can be detected on a PET (positron emission tomography) scan. We do a lot of PET scans of patients with cancer using an injected radioactive form of glucose labeled with 18F, 2-deoxy -2-[18F]fluoro-D-glucose, or FDG for short. Cells take up FDG but do not metabolize it and we can see on a PET scan where the glucose-avid cells are. PET scans with FDG work extremely well in cancer precisely because of the Warburg effect — i.e. many cancers depend on glucose for their fuel. In Fig. 1 (left, below) the scan shows a small metastasis in the liver, just above the kidney; Fig. 1(right) shows same patient now with many new liver metastases and one new spot in the lung.

Figure 1: Metastatic Cancer from a primary tumor in the colon. The primary cancer in the colon is not visible as it was removed surgically, but the disease had already spread microscopically so that by the time of the PET image on the left there’s a metastasis in the liver, and the right image shows multiple liver mets and one small lung met. (Note that darker means more FDG uptake.)
The heart and brain normally use a lot of glucose, so they “light up” with FDG, and, due to FDG excretion, kidneys and bladder are also seen well. There’s normally only mild background uptake detected in other tissues. Intense uptake in an unexpected location strongly suggests a primary cancer or a metastasis. For this reason, PET has become very useful in the management of many cancers, where it can show tumors clearly and can demonstrate the effects of treatment. My early interest in the Warburg effect was obviously fueled by what PET scans could show us.
A word of caution: Cancers resist overly broad generalizations. Notice that I always say “many cancers” and not “all cancers.” Otto Warburg made a very important observation, but he went a little too far by stating that persistent glycolysis was the common feature of all cancers. The observation holds up well for many aggressive malignancies but by way of counterexample, 80% of prostate cancers are not especially aggressive, nor are they avid for FDG (or glucose), i.e. the Warburg effect does not apply.
Back to our study idea: We realized that gluconeogenesis and release from glycogen stores would prevent blood glucose concentration from falling to a level low enough to starve cancer cells. However, it became clear that by reducing insulin signaling, dietary CHO reduction would cause many effects which were known to inhibit cancer growth. Some of these were systemic, such as ketosis triggered at CHO restriction to less than 50 grams/day. Ketosis had been reported to inhibit cancer growth in cell culture studies in our lab and others (1,2), animal cancer studies (3,4) and a case study of two children with brain tumors (5). Other expected effects are changes in all the intracellular signaling molecules downstream of the insulin receptor, which regulate their growth, proliferation, and resistance to apoptosis (cell death signals known in all cells), etc. In the last blogpost, Richard described an animal study which demonstrated insulin’s involvement in downstream signaling and response to diet in cancer.
Cancers are now being treated with drugs that individually target these intracellular signaling molecules that are controlled by insulin. These new drugs have shown some efficacy but are often limited by side effects due to the drug interactions with normal tissues. Normal tissues, however, are tolerant of the effects of reduced insulin signaling. This is apparent from the safety of low CHO diet investigations in overweight people and people with diabetes as well as healthy subjects of normal weight. It seemed reasonable to us that a low CHO insulin inhibiting (INSINH) diet could target the same molecules as the drugs and could plausibly inhibit or even kill cancer cells, but would be safe for normal tissues. So, if a study showed safety/feasibility as well as some evidence for efficacy, it would open the door for further investigation of this diet at least as an adjunct to drug therapies. In short, an INSINH diet, by systemic and synergistic effects on multiple signaling molecules, might eventually be shown to reduce drug doses and therefore side effects, boosting efficacy at the same time. What was most surprising to us was that nobody had done this before.
Our goal was to implement a ketogenic INSINH diet for 28 days to see if this diet was safe and feasible in cancer patients and to test for efficacy using the change in FDG uptake on a PET scan. We were striving for ketosis, i.e. the strictest type of low carb diet, with the thought that we might engage/recruit as many cancer inhibitory mechanisms as possible. Diets are hard, but almost anyone can stay on a diet for a month—and we wanted the patients to have a chance to succeed. Furthermore, PET scans are very sensitive: some cancers show changes due to treatment as early as a week, so a month might even permit us to see evidence of improvement on the scan.
What about the results? Ten patients is too small a sample to draw firm conclusions. And it wasn’t so easy after all to get ten sick patients to do this trial. Four of the patients continued to have progressive cancer by our follow-up PET scan at one month, while five patients showed stable disease and one, a partial remission. We’d agree that these findings aren’t so remarkable in themselves. But the details are much more interesting: The patients with the worst PET scan results at study’s end were principally those who had the least degree of insulin inhibition, i.e. the least amount of ketosis (only five times their baseline level); whereas those that had the best PET scan results were those that had the most insulin inhibition, or the most ketosis (17 times baseline)—see A below (where β-hydroxybutyrate is the ketone body we measured); asterisks represent a significant difference (p<0.02). Figure B confirms a general inverse relationship between the extent of ketosis and insulin concentration, expected from CHO restriction (p<0.03).

In conclusion the best metabolic response to the INSINH diet gave the best PET scan response, the worst metabolic response gave the worst, but calorie reduction and weight loss did not demonstrate a measurable relation to PET outcome.
Our trial might be viewed as an unremarkable pilot study with too few patients to draw many inferences. But that would miss the point. We think what’s more important is that we may have opened a door, long overdue, to systemic study of dietary compositional change in cancer therapy– an insulin inhibiting diet is now worth a further look!! We hope there will now be the opportunity to study INSINH diets in more patients; to see if standard therapies and newer drug treatments can be improved by dietary adjunctive therapy; to find biomarkers to pre-identify which patients are/aren’t likely to benefit from diet; to tease out the effects of calorie restriction from those of carbohydrate restriction.
The next post will describe in more detail the underlying hypothesis about carbohydrate restriction/insulin inhibition as a potential therapy for cancer.
References
1. Demetrakopoulos GE, Brennan MF. Tumoricidal potential of nutritional manipulations. Cancer Res. 1982;42(2 Suppl):756s-65s.
2. Magee BA, Potezny N, Rofe AM, Conyers RA. The inhibition of malignant cell growth by ketone bodies. Aust J Exp Biol Med Sci. 1979 Oct;57(5):529-39.
3. Mavropoulos JC, Isaacs WB, Pizzo SV, Freedland SJ. Is there a role for a low-carbohydrate ketogenic diet in the management of prostate cancer? Urology. 2006 Jul;68(1):15-8.
4. Moulton CJ, Valentine RJ, Layman DK, Devkota S, Singletary KW, Wallig MA, et al. A high protein moderate carbohydrate diet fed at discrete meals reduces early progression of N-methyl-N-nitrosourea-induced breast tumorigenesis in rats. Nutr Metab (Lond). 2010;7:1.
5. Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995 Apr;14(2):202-8.
Targeting insulin inhibition as a metabolic therapy in advanced cancer. Part 2. The hypothesis.
Posted: November 30, 2012 in Cancer, Cell Signaling, evolution, ketogenic diet, low-carbohydrate dietTags: biochemistry, Cancer, carbohydrate, evolution, hunter-gatherer, insulin
Last time I discussed our pilot study showing the effects of carbohydrate (CHO) restriction & insulin inhibition (INSINH) in patients with advanced cancers. We described how the molecular effects of INSINH plus systemic (total body) effects like ketosis might inhibit cancer growth. My goal now is to present the underlying hypothesis behind the idea with the goal of understanding how patients with cancers might respond if we inhibited insulin’s actions? Should all patients respond? If not, why not? Might some patients get worse? These ideas were described briefly in our publication describing our pilot protocol.
The hypothesis hinges on understanding how natural selection works, and how it works on two levels. The first is based on our evolutionary heritage and how humans evolved biochemically. The second level addresses how cancers evolve within people. In sum, we’re considering an intersection of human evolution over our long prehistory vs. cancer evolution over several years within human beings. Let me explain:
Human evolution: We’ll arbitrarily date the first humans from the evolution of Homo erectus about 1 million years ago, or from Homo sapiens, approximately 200,000 years ago. Since agriculture emerged about 10,000 years ago we can conclude that our ancestors were hunter-gatherers for at least 95% of our existence. According to paleontological evidence dietary CHO of hunter-gathers was in the range of 20-35% of total caloric intake. For most of human evolution we had no bread on the table, nor cakes, puddings, pies or chips. Wild fruits gathered during hunter-gather days don’t measure up to the huge domesticated fruits now abundantly available all year round. It’s difficult to imagine how humans didn’t starve during the winters in northern Europe or on the Asian steppes. Even in Africa, the cooling climate beginning around 100,000 years ago forced migrations of people toward more hospitable (and food abundant) climes. At the very least, there’s little doubt that our forebears did not consume excess dietary sugar and starch resulting in obesity and metabolic syndrome. In short, for 95% of human existence, the paleontological record shows that we were very tolerant of minimal CHO intake.
But the best evidence is in our biochemistry: under well fed modern conditions our brain uses about 130 grams of glucose/day (for most of us in the developed world). During a fast our brain continues to use glucose derived from the breakdown of glycogen in our liver as well as from glucose synthesis (gluconeogenesis) largely from body proteins. However, the continued consumption of our own proteins for a fast lasting longer than a few days isn’t a good evolutionary survival strategy as the protein would be stolen from our muscles, killing us rapidly from heart and diaphragm failure.
A key survival strategy that allowed conservation of body protein — the ability to generate ketone bodies as an alternative fuel source — is what actually survived. Under conditions of fasting—total reduction in nutrients (including carbohydrate) — glucose concentration will fall in the bloodstream thereby causing less pancreatic insulin secretion. Breakdown of fat in fat cells (lipolysis) is ordinarily inhibited by high levels of insulin. Conversely, fat breakdown accelerates under conditions of fasting (low insulin signaling), causing release of fatty acids (FA) into the blood. FA’s circulate to the liver where they then get metabolized into circulating ketone bodies (KB). That’s why fasting results in ketosis, as does strict low carbohydrate dieting (e.g. less than 50 grams of CHO/day). The brain uses 130 grams of glucose/day under our ordinary modern dietary conditions. But during starvation/fasting, over a 6 week period, the brain switches to ketone bodies to supply more than 2/3 of its fuel. This masterful metabolic switch spares protein breakdown and permits humans to tolerate starvation as long as fat stores hold out. That’s why people can survive without food sometimes for months, whereas absolute water deprivation can kill in just a few days.
One way to describe the evolution of our metabolism is that, over millennia, environmental conditions selected those individuals for survival who a) stored up enough fat during the more plentiful months to provide an energy source for their body and b) had the enzyme capacity to make ketone bodies (KB) from the stored fat and could utilize these KB as brain fuel to reduce the brain’s need for glucose derived from protein.. These individuals survived over the long lean winters to reproduce, and they passed on the genes that enabled this capacity.
Anyway, civilization and agriculture changed the availability of many foods, but especially grains. Bread became the ‘staff of life.’ In recent years, excessive, cheap CHO, coupled with the Food Pyramid’s recommendations to consume CHO to the tune of 55-70% of total caloric intake have likely contributed to widespread metabolic syndrome, obesity, type 2 diabetes, dyslipidemias and hypertension. Finally, obesity and high insulin concentrations in the blood (both linked to dietary CHO excess) have now been associated with an increased risk for a variety of different cancers.
Obesity has in fact surpassed smoking as the greatest behavioral risk factor for cancers in the U.S.
Reducing cancer risk by CHO restriction would seem to be a meaningful preventive cultural dietary goal. The idea appears to resemble our study where we treated cancers with a VLC/INSINH diet, but it’s not quite the same thing. Cancer cells behave differently from normal cells, so, metabolically, treatment isn’t quite the same as prevention.
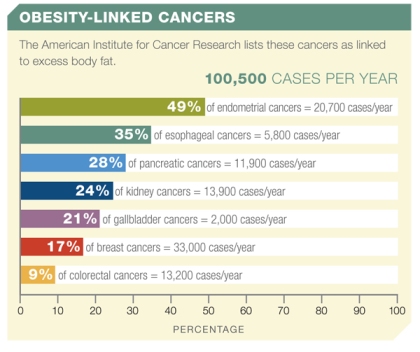
Figure 1: The American Institute for Cancer Research links more than 100,000 new cases of cancer per year to obesity.
Regardless of the initial insult, each one of these abnormal but not yet malignant cells must evolve over time within a human organism. We must remember that mutations in normal cells happen all the time and they are usually maladaptive or destructive to the cells; healthy cells have mechanisms to protect the organism by a kind of cell suicide or apoptosis, best thought of as self-pruning of maladaptive cells. So most mutations within these aberrant cells will cause cell death and will not result in a cancer. But there are mutations, seen in many types of cancers, which can cause resistance to apoptosis and therefore permit abnormal cell growth to continue.
This is where the evolution of humans and the evolution of cancers within humans collide. In the modern developed world most cancers evolve within individuals under conditions of substrate/nutrient abundance, and especially CHO abundance. And while famine continues to plague people during wars and in underdeveloped regions, it’s nevertheless reasonable to suggest that sustained ketosis and INSINH are not prevalent in the modern developed world. Therefore in the developed world, cancers in many people are unlikely to have experienced a microenvironment due to the metabolic effects of insulin inhibition (INSINH) or ketosis (a limiting state of INSINH).
In summary, INSINH would represent a new metabolic selective pressure for many cancers to which they would reasonably be vulnerable.
Hypothesis: Humans are adapted to starvation and the metabolically related low CHO diet/INSINH state. On the other hand, cancers in large cohorts of people in developed countries are under high chronic insulin stimulation and are largely unexposed to the unfamiliar INSINH state. These cancers will sensibly express a wide range of molecular and metabolic vulnerabilities to INSINH. However, they may express an equally wide range of adaptive mutations. Investigators have confirmed both vulnerability of some cancer types to inhibition by added ketone bodies as well as continued uninhibited cancer growth with added KBs in other cancers.
Two important corollaries about cancers in hunter-gatherers and modern ‘low carbers’:
First, there’s no a priori reason that cancers can’t arise in individuals on INSINH/VLC diets in the modern world, or in modern (or prehistoric) hunter-gathers. Cancers initiate from a series of mutations due to causes that may be intrinsic to cell metabolism or to the external environment. Even for hunter-gatherers, heavy metals in ground water (even without civilization), background radiation all around us, smoke from smoking tobacco or herbs, campfires and cooking (hunter gatherers had controlled fire for close to 1 million years) and chronic inflammation are some of the factors that have existed for millennia, even if more pervasive since the onset of civilization. Although anecdotal and paleontological evidence is all that we have, cancers have been reported in ancient bones. Nothing would prevent cancers from forming, even in the likely reduced insulin signaling state of early hunter-gatherers (or in modern people on VLC/INSINH diets).
Second, while fewer cancers might be expected to develop, those hunter-gatherers (or modern low-carbers) who do develop cancers would not experience a VLC diet as a new selective pressure. Our pilot study, in fact, excluded patients who had been on a VLC diet within 3 years of the diagnosis of their cancer because a VLC/INSINH diet would not be expected to have any positive effect on their cancers.
To sum up: For people on high CHO diets who develop cancer, a low carbohydrate diet targeting insulin (VLC/INSINH) may be therapeutic. The rationale is that control of insulin and the presence of ketone bodies provides a new selective evolutionary pressure to which the cancers may not be adapted.
On the other hand, those cancers that develop in people who are already on low-carbohydrate diets (already in a state of dietary insulin inhibition) will not be expected to be vulnerable to the VLC/INSINH diet.
About these ads